
Infectious Disease Surveillance
Detect known and novel pathogens to accelerate outbreak investigations.
The COVID-19 pandemic has brought intense awareness to zoonotic diseases and the real danger they pose to global human health and welfare. The pandemic highlights how easily an infectious agent can spread through a population, particularly when many affected individuals are asymptomatic or have symptoms easily mistaken for any number of diseases. As such, a sensitive and specific means of detecting the virus has been essential to tracking it and implementing effective public health measures.
A single assay that can provide information on viral genomic data, microbiome composition, information on co-infection, and the transcriptional status of the host immune response would be powerful. Common samples collected to evaluate SARS-CoV-2 are complex samples like saliva and nasopharyngeal swabs which contains a mixture of bacterial and human cells.
Enhance detection sensitivity with complete COVID genome coverage and help labs find more information while reducing sequencing costs.
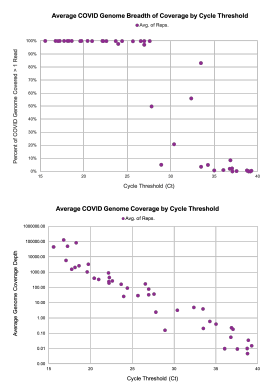
CRISPR-powered rRNA depletion improves virus strain calling, boosts the number of human genes detected above noise and increases detection of bacterial species.
With DepleteX, you can gain a deeper view of expression profiles of individual cells.
Shotgun metatranscriptomics sequencing is an invaluable tool to understand the constantly evolving virus and pathogens by offering unbiased data. However, much transcriptomic data can be uninformative, as noise from overabundant ribosomal RNA (rRNA) sequences from both human and bacterial cells drown out the signal from important transcripts.
“The problem [with NGS] is that a lot of the stuff… is not what you’re looking for — it’s more noise. And most of it will just be human DNA, because it’s taken from a human. What one would want to do is find the stuff that’s not human, that is… the DNA sequence reflecting the pathogen, and somehow get rid of all the noise. And that’s what Jumpcode’s technology can do.”
Nicholas Schork, Deputy Director and Distinguished Professor of Quantitative Medicine
Acquire viral genomic data, microbiome composition, information on co-infections, and host-immune response in a single workflow.
Explore featured resources
CRISPRclean Plus Stranded Total RNA Prep with rRNA Depletion
CRISPRclean Plus for SARS-CoV-2 Shotgun Metatranscriptomics
DepleteX® Nasopharyngeal Microbial RNA Boost
Connect with us to learn more
For the US patent, the patent number is US 10,604,802 entitled Genome Fractioning. The patent publication is available here.
For the EP patent, the patent number is EP3102722 entitled Genome Fractioning. The patent publication is available here.