Single Cell Sequencing

Gain a deeper view of single cell expression profiles with DepleteX™.
Transcriptional profiling has been revolutionized with high throughput, massively parallel, single cell RNA sequencing. Microfluidic technologies produce comprehensive data sets that allow investigators to understand complex cell mixtures, identify cell types present in healthy and diseased tissues, and create cell type specific transcriptional signatures. These technologies are dramatically enhancing our ability to identify transcriptional and cellular perturbations driving disease and basic biology understanding at the individual cell level.

Uninformative transcripts dominate sequencing reads.
One problem faced by single cell RNA-seq methods is that greater than 90% of single cell RNA-seq dataset is noise and uninformative1. While computational algorithms have evolved to parse out the true signal, commonly available microfluidic technologies enable only sparse sampling of RNAs from each cell, with many genes represented by a few sequencing reads. This limit is partially due to an abundance of biologically uninformative RNAs which dominate sequencing reads and limit detection of the moderately expressed transcripts that often drive biological differences between cell types.
What if there was a simple solution that removes uninformative reads in-vitro, thereby redistributing sequencing reads to unique, biologically relevant transcripts?
Jumpcode’s Solutions are Compatible with Multiple Technologies
10x Visium Spatial Sequencing
Depletion identifies two additional clusters with biologically relevant lowly expressed markers in spatial transcriptomics ovarian tissues.
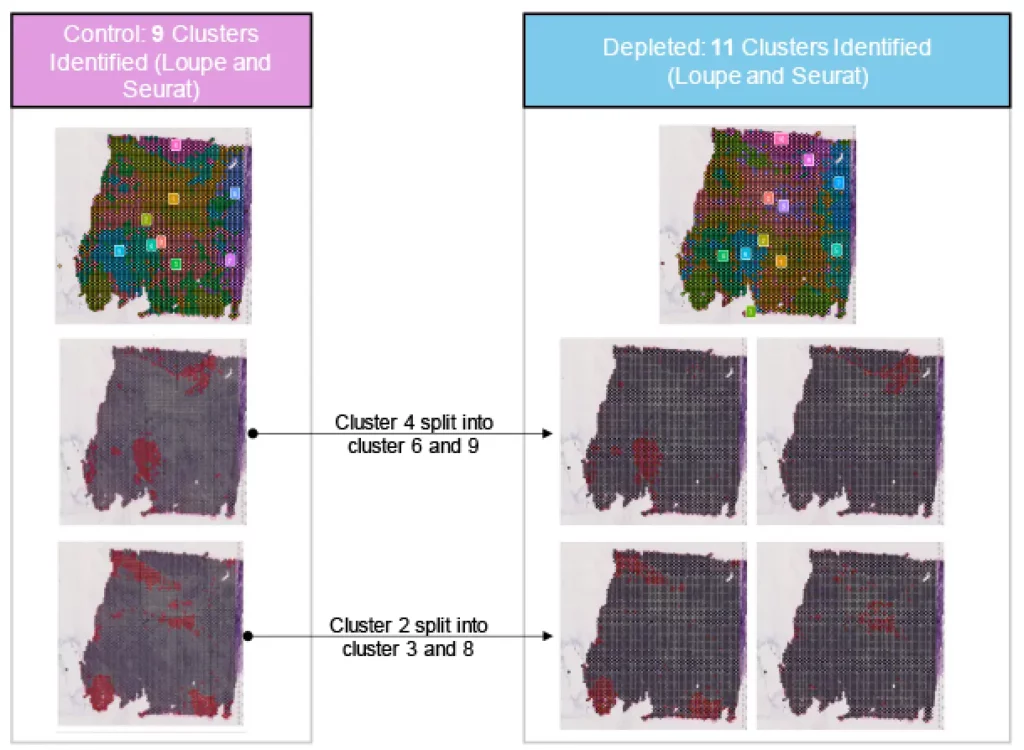
~52% of reads within ovarian tumor was redistributed after depletion using Jumpcode depletion. This nearly doubles the percent of reads aligning to the transcriptome from 40% to 70%. Depletion increased statistical confidence in identifying spatial, biologically relevant genes.
PacBio MAS-Seq
Jumpcode-depleted data identifies four additional rare cell clusters in PBMC
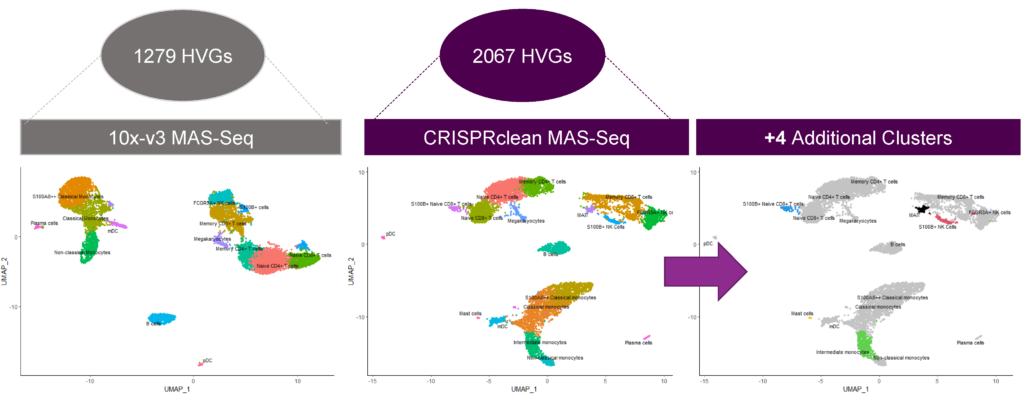
Depleted samples have 4 additional clusters under the same analytical conditions as the control because of the increased detection of highly variable genes (HVGs).
BD RhapsodyTM Whole Transcriptome Single Cell Sequencing
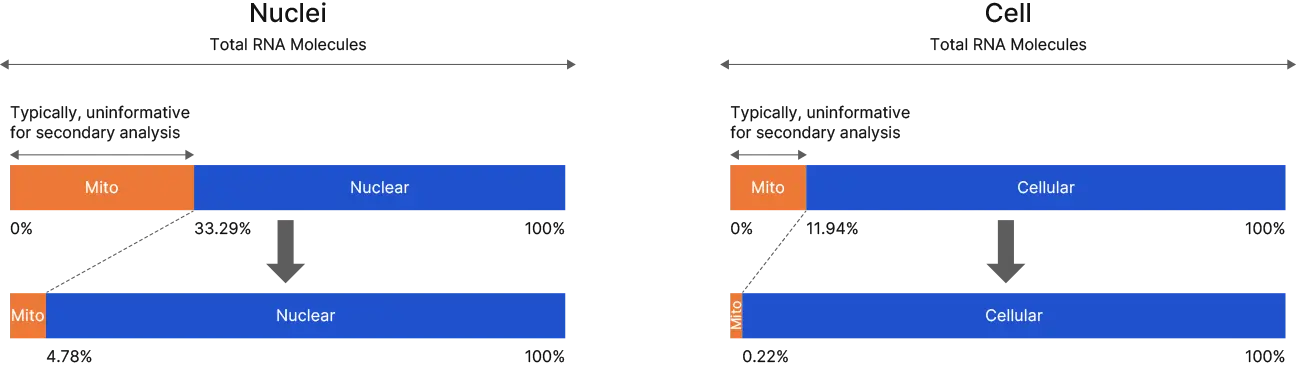
Remove mitochondrial interference and enhance single cell RNASeq data through Jumpcode DepleteX™ and the BD RhapsodyTM System:
To see the portfolio of programmable endonuclease enzymes click here for the Revvity product page.

Connect with us to learn more
1 Qiu, P. Embracing the dropouts in single-cell RNA-seq analysis. Nat Commun 11, 1169 (2020).
For the US patent, the patent number is US 10,604,802 entitled Genome Fractioning. The patent publication is available here.
For the EP patent, the patent number is EP3102722 entitled Genome Fractioning. The patent publication is available here.